How to use "MPDyn2"
Contents of the "MPDyn2" package
You can extract the following subdirectories from distributed archive file,
"MPDyn2.tar.gz".
tar -zxvf MPDyn2.tar.gz
| MPDyn2/ |
main directory |
| MPDyn2/bin/ |
executable files |
| MPDyn2/param/ |
parameters |
| MPDyn2/source/ |
source codes |
| MPDyn2/case_study/ |
sample (initial configuration and input files for test calculations) |
Building MPDyn executables
For an easy compiling and building the MPDyn executable, a set of makefiles
is provided in the ./MPDyn/source subdirectory. Distributed makefiles will
work for the following computer.
-
- Makefile.gnu : Gnu Compiler (gfortran)
- Makefile.intel : Intel Compiler
For example, one can make the executable with the intel compiler ver.15.*
on Intel Xeon machine with
make -f Makefile.intel
in the directory ./MPDyn/source. Then, MPDyn executable "MPDynS"
will be built in the directory ./MPDyn/bin. This file is the executable
on a single cpu. The executable file for MPI-parallel computing, MPDynP,
is created by using the flag, MPI;
make -f Makefile.intel MPI
IMPORTANT:
Makefiles provided here presume MPI library correctly installed prior to
building MPDyn. If one uses only a single node computer without MPI library,
the line starting from "FC=" in the Makefile should be edited.
If this line says "FC= mpif90", then one should rewrite it "FC=
(compiler command)"; for example, in case of Makefile.intel, it should
be "FC= ifort".
In addition to the simulators, several useful tools are also provided in
this package;
- MPDyn_Prep : make an initial configuration in the CRD format
- PSFMaker : make a PSF file (for CHARMM)
- Eplot : convert monitor files into several files with the plotter-friendly format.
- Xconverter: converter for the restart-configuration file (restart.dat)
- TransCell : rotate the system (basic cell) to set the a-axis on the x-axis
and the b-axis on the x-y plane.
- ToolParam : make the molecular coordinates for reference rigid-body
To build these tools, one should type
Make -f Makfile.intel7 TOOLS
in the directory ./MPDyn/source.
Data flow during MD calculations
MPDyn enables us to perform several kinds of molecular simulation with appropriate inputs. Here, for example, requested input files for an MD simulation as well as the relevant output files are listed.
| input |
MPDyn |
output |
User files
(Initial configuration)
initial.crd (ascii)
(Job script file)
input.data (ascii)
(PSF file / CHARMM only)
*.psf
Force Field Database
(Ex. CHARMM)
./param/top_charmm27.prm
./param/par_charmm27.prm |
-> |
MD
HMC
PIMD
CMD
DPD
Analysis |
-> |
(Final configuration)
restart.dat (binary)
final.pdb (ascii)
(monitor files)
jobname.moni
(trajectory files)
jobname.r001
(velocity files)
jobname.v001 |
Input files
Original files
The following three files are required to start the simulation.
- Job script / condition file ("input.data")
- Initial configuration file ("initial.crd")
- PSF file (CHARMM protein structure file, "***.psf")
Note: A PSF file is necessary only when the CHARMM force filed is used.
Common parameter files
- parameter file ( CHARMM / OPLSAA / BKS)
- topology file ( CHARMM / OPLSAA /BKS)
Note: One can use the EAM (Embedded Atom Method) potential to treat the
metal. In this case,
How to prepare the input files
- Job script (condition file )
Filename : "input.data" (several samples are available in the
Tutorial section.)
This file is always needed to set the simulation condition.
- Initial configuration file
File name : "initial.crd" and "initialRB.dat" (the
latter is needed when one uses a rigid-body model)
There are several ways to prepare the initial configuration for simulations.
The file, "initial.crd", should be prepared in the CRD format
of the CHARMM (.crd).
- A configuration file of several types of format such as a PDB file can
be converted in to a CRD file by using "InsightII" or "CHARMM"
package. These programs are also useful to build the model system and to
generate a PSF file. The usual CRD file does not contain any information
on the size of simulation box. To carry out the MD calculation in the periodic
boundary condition, it is needed to define the cell size. MPDyn requests
the following three lines, which describe the cell matrix, in the last
of the CRD file.
ax bx cx
ay by cy
az bz cz
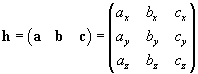
Finally, the name of the crd file must be changed into "initial.crd".
For molecular simulations, this is one of the easiest way to start the
simulation.
- "MPDyn_Prep" can be used to convert a PDB file into a CRD, "initial.crd".
However, it should be noted that, in this program, it is assumed the atom
and residue names are correctly assigned in the original PDB file. If this
condition is not met, "MPDyn_Prep" will not work correctly. To
satisfy this condition, it is sometimes needed some text-edit of the PDB
file. To build the initial configuration for the rigid-body system,"initialRB.dat",
this program is quite useful. This program require a file, "input_prep.data".
In this file, the following information should be written in the order.
1. Force Field [ CHARMM, OPLS ]
2. The name of the parameter file [Just put "D" for the use of
default parameter.]
3. The name of the topology file [Just put "D" for the use of
default file]
3. Whether rigid-body model is used or not. [ .T. / .F. ]
4. The number of molecular species in the system
5. Molecular names ( should be written for all species in one line. )
6. The numbers of molecules (the same as above)
7. The numbers of atoms per molecule (the same as above)
(The following lines are needed only when the rigid-body model is used with the CHARMm force filed.)
8. Blank or comment line
9. The status (file format) of "restart.dat"
10. thermostat [ ON / OFF ], the Nose-Hoover chain length (only in case
of ON)
11. barostat [ON / OFF ]
12. Whether use periodic boundary condition or not. [ .T. / .F. ]
13. PSF file name |
NOTICE: FOR A LARGE SYSTEM
An important change has been made in the file format of "initial.crd"
only for a system containing more than 100,000 atoms. If a user has such
large number of atoms, MPDyn assumes that the "initial.crd" is
prepared with the following format.
Initial few lines starting from asterisk * or blank lines are regarded
as comments as usual. First meaningful line is total number of atoms written
in '(i10)' (I use the fortran command for the format). Each following line
contains "atom number", "residue number", "residue
name", "atom name", and "coordinate (X, Y, Z)"
for each atom. The new format is '(2i9,2(x,a4),3f12.5)', though the original
crd format was '(2i5,2(x,a4),3f10.5)'. After these lines, the simulation
box size should be given when user wants to use periodic boundary conditions.
- PSF (structure file)
A PSF file is required, when one start the simulation with the CHARMM force
field. There is several ways to produce PSF files. Again to use "InsightII"
is the easiest way to do so. However, for the MD calculation of the system,
in which the molecule that is not registered in InsightII originally contains,
user has to make the structure data in other way. If the academic CHARMM
program is available, it will be quite useful to make the PSF files.
An alternative way to generate a PSF file is to use "PSFMaker",
which is supplied as a tool program in this package. However, the "PSFMaker"
does not work for the system containing molecules described multi residues,
e.g. proteins. This program requires some inputs. , the following information
should be written in the following manner.
1. The name of the parameter file [Just put "D" for the use of
default parameter.]
2. The name of the topology file [Just put "D" for the use of default file]
3. The name of PSF file
4. The number of molecular species in the system
5. Molecular names ( should be written for all species in one line. )
6. The numbers of molecules (the same as above)
7. To finalize this script, add "<end>" in the last line.
|
- Force Field data
Several files containing force filed (FF) data are supplied with the program
package. For each force field, a pair of files, i.e., topology and parameter
files, is required to assign the FF parameter correctly. The CHARMM format
is adopted for these files.
- CHARMm : We can make use of the original CHARMM FF parameter files, which is downloadable form the www homepage:
http://www.pharmacy.umaryland.edu/faculty/amackere/force_fields.htm
Detailed information on the format of the topology and parameter files
are supplied by the CHARMM www site, and a short comment is also available from here.
- OPLS : The parameter set for the OPLS-AA will be provided. Currently available
file is only for a few water model. Whenever one try to use this for some
other molecules, some edittings of the topology and parameter files are
needed. A set of these files are provided in the CHARMM web site by Brooks
and his co-worker.
- BKS : This parameter set is used for inorganic materials such as silica.
A set of parameter files is available. Note that, in this FF, non-bonded
interaction is described by Exp-6 + Coulomb. For Exp-6, no combination
rule is used.
- EAM : For embedded atom method (EAM), neither topology nor parameter files is used. The interaction parameter is prepared as a table function for each pair of atoms. For example, Ni.1, Ni.2, Ni-Ni.3 are required for the Ni crystal system.
- Force Field data for DPD
A file which contains the lists of the parameter, a, for the conservative
force is needed.
Fc = a ( 1 - (r/rc)n )m
(n, m = 1; the conventional form)
This file should be written in the following way;
The number of atom types, n
Blank line
a(1,1) a(1,2) ... a(1,n)
a(2,1) a(2,2) ... a(2,n)
a(n,1) a(n,2) ... a(n,n) |
n and m are specified by the name of the force field.
How to execute MPDyn
You will have two executables, MPDynS and MPDynP. The former is just for
single node calculation and the latter is for multiple nodes calculation
by using public domain MPI library.
To start MPDyn on a single node computer, just type
"MPDynS"
For parallel computing, you may use,
"mpirun -np ** MPDynP"
** is the number of nodes you want to use. However, the way to execute
a mpi job highly depends on the computer system, you should check it before
submitting your job.
Other tools
Eplot
"Eplot" converts the monitor files (*.moni) into several files
with the plotter-friendly format. This needs some input data including
the simulation method and ensemble used in the simulation as well as the
filenames, which are to be converted. Before executing "Eplot",
make a directory, "./E_data", where the converted files are stored.
Xconvert
"Xconvert" is a tool to convert the final configuration file
of the previous MD calculation, "restart.dat", into a different
configuration file, which is to be used in the successive MD run with some
condition being different from the previoud MD. For example, you may need
to change the thermodynamic ensemble on the way of an MD simulation. In
such case, "Xconvert" helps you making a new "restart.dat"
without changing atomic velocities. You can also use "MPDyn_Prep"
for this purpose, if and only if you do not care that the atomic momentum
are completely refreshed. The resultant (converted) data are given in the
file, "restart.converted". This is also useful for rigid-body
models.
"Xconvert" needs two input files, "input.data" and
"input_convert.data". The former should be the same file that
is used in the previous MD except that the flag 'STATUS' should be switched
to 'Restart', and the latter is the script file where only the condition
to be changed is written in the same format as the job-script file, "input.data".
"input.data" is used to get the information before changing the
condition, while "input_convert.data" is used to get the new
condition. The readable flags in "input_convert.data" are "METHOD",
"PRESSURE", "TEMPERATURE", "PI_PARAM", "CMD_PARAM",
and "CONSTRAINT". To switch off the barostat or thermostat, you
should select "METHOD= OFF" in the "PRESSURE" or "TEMPERATURE"
keywords (see jobscript section). A flag, "BATHREFRESH", is also
available, which is used to initialize the variables of barostat and thermostats.
"TIMEREFRESH" is used to initialize the simulation time (t=0).
To change the data format of restart.dat, (i.e., binary --> ascii OR
ascii --> binary), the flag "CHANGE_FORM" can be used. Given
the flag, the data format of the genrated "restart.converted"
should have been converted.
Usage: you may put the following lines in "input_convert.data".
>> CHANGE_FORM
<<
We also have two more keywords for Xconvert; "EXTENSION" and
"MOVE". "EXTENSION" is useful when you duplicate the system for the next
simulation, while "MOVE" gives you a moved (translcated) restart
file. Both keywords has three options, "X=","Y=","Z=".
For "EXTENSION", you have to put integer numbers for the options,
giving the number you want to duplicate the system for each direction.
In case of "MOVE", this is just a dislocation vector, which should
be real number. e.g.,
>> EXTENSION
X= 2
Y= 2
<<
This gives you a restart.converted having a 4 times bigger box: 2x2x1 copy of the original simulation box.
Tips for Xconvert:
Xconverter may work when you change
1. simulation METHOD from MD -> PIMD or CMD
2. bath control / turn on or off baro and thermostats
3. data format of restart.dat. (e.g. ascii/binary)
When you want to change the simulation method from PIMD/CMD/HMC to MD,
you may use final.pdb to make initial.crd by using MPDyn_Prep. When you
want to relax any holonomic constraints in the system, any convertment
is required for "restart.dat".
ToolParam
"ToolParam" is an optional tool, which generates the coordination
of rigid-bodies in the body-fixed frame in "RigidBodyModel.prm"
in the ./param directory. It is necessary to some additional information
in the topology file, when this program is used. The source code of this
tool is "MakeBodyFixedCoordinate.f90". It may be instructive
to see this source code to make a "RigidBodyModel.prm".
Return to HOME