

Virus Capsids/
Virion ModelingMolecular Simulation
Virus capsids
Molecular-level modeling of virus capsids remains a highly challenging task due to the many factors that contribute to capsid stability and are still not fully understood. Virus capsids constitute one of the key targets of our molecular modeling and simulation studies. Using the hepatitis B virus (HBV) capsid as a model system, we have constructed a full capsid model containing pregenomic RNA (pgRNA). We have performed simulations using both all-atom and coarse-grained force fields to investigate the structure, stability, and formation mechanisms of the HBV capsid.
The extended coarse-grained SPICA force field has also been applied to an empty poliovirus capsid, demonstrating a highly stable and rigid capsid structure in CG-MD simulations over microsecond timescales. Furthermore, the HBV capsid containing pgRNA was successfully simulated using the SPICA force field. CG-MD simulations were also employed to investigate the adsorption process of the reverse transcriptase inhibitor entecavir into the HBV capsid.
 |
 |
| HBV capsid model in all-atom MD. Capsid proteins are shown in grey and RNA is shown in yellow. | Simulated poliovirus capsid with the SPICA force field. |
Virion modeling
Hepatitis B virus (HBV) particles are formed through the budding of preassembled cytoplasmic nucleocapsids into endoplasmic reticulum (ER) membranes containing viral envelope proteins. Understanding the molecular organization and dynamics of the resulting enveloped virion remains a major challenge, as detailed molecular-level information on capsid–envelope interactions is still limited.
n our recent work, we have developed a molecular model of the complete HBV virion and performed coarse-grained molecular dynamics (CG-MD) simulations to investigate its structural and dynamical properties (Urano & Shinoda, J. Phys. Chem. Lett., in press). Using a chemically realistic residue-based coarse-grained model with the SPICA force field, we constructed HBV envelope systems both with and without the icosahedral capsid and compared them with a pure lipid vesicle.
Our simulations reproduce experimentally observed electron density profiles of HBV virions and reveal stable molecular interactions between capsid spikes and the transmembrane regions of HBV S proteins. These interactions involve not only experimentally known contact sites but also previously unrecognized residues that contribute to capsid–envelope association. The results demonstrate that protein–protein interactions play a dominant role in stabilizing the virion structure, whereas lipid-mediated interactions are comparatively weak and transient.
We further found that lipid diffusion within the viral envelope is significantly slower than in a simple lipid vesicle, and that the presence of the capsid further suppresses lipid mobility in the inner leaflet. This highlights strong protein-mediated modulation of membrane dynamics in enveloped viruses. Despite the absence of M and L proteins in the current model, the simulations suggest that essential features of the HBV envelope–capsid complex can be captured by S proteins and lipids alone at this scale.
This study provides the first molecular dynamics simulation of a viral particle containing both the capsid and the surrounding lipid–protein envelope, offering a molecular-level view of the HBV capsid–envelope complex. Our current efforts are directed toward extending this model to include additional viral proteins, nucleic acids, and diverse lipid components, with the goal of constructing a more complete and realistic HBV virion model.
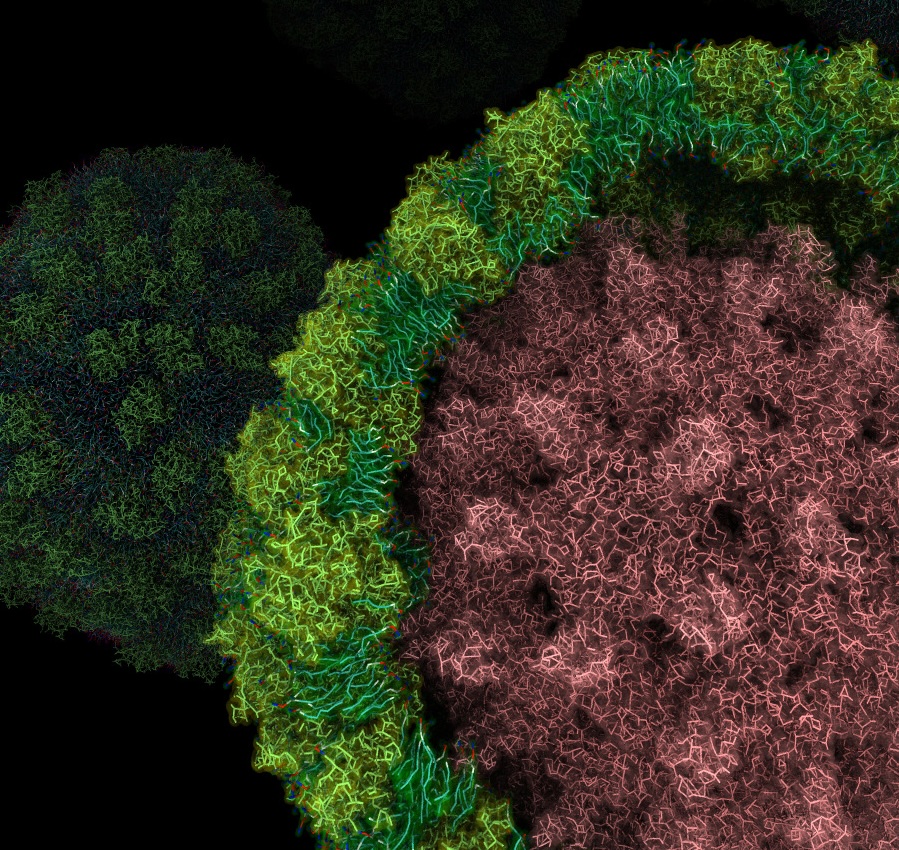 |
| Cross-sectional view of a simulated hepatitis B virus (HBV) particle. The pink region represents the capsid, surrounded by a lipid–protein envelope composed of S proteins (yellow–green) and lipid molecules (cyan), illustrating the molecular organization of the HBV envelope. |