

Electrolytes for batteryMolecular Simulation for battery
Polyeletrolytes (PFSA membrane)
Proton exchange membrane fuel cells are a highly efficient and green alternative power source. The fuel cells currently use perfluorosulfonic acid (PFSA) ionomers as proton exchange membranes due to their high proton conductivity and high mechanical stability. To investigate the effect of PFSA structure on the proton conductance or gas leakage, a series of molecular dynamics (MD) studies have been done. All-atom MD is successfully used with rather short PFSA ionomers. For an application for fuel cells, long chained PSFA polymers( > 120 monomer units) should be used. We developed quantitative coarse-grained (CG) model for PFSA using the hybrid IBI/SDK model, which is the combined interative Boltzmann inversion (IBI) technique and the SDK (SPICA) force field, and applied to PFSA membranes with long polymers. After relaxation CG-MD run for microseconds, we used reverse mapping technique to reconstruct the membrane system at all-atomic level. The technique works well for PFSA membranes with 125 monomer length.
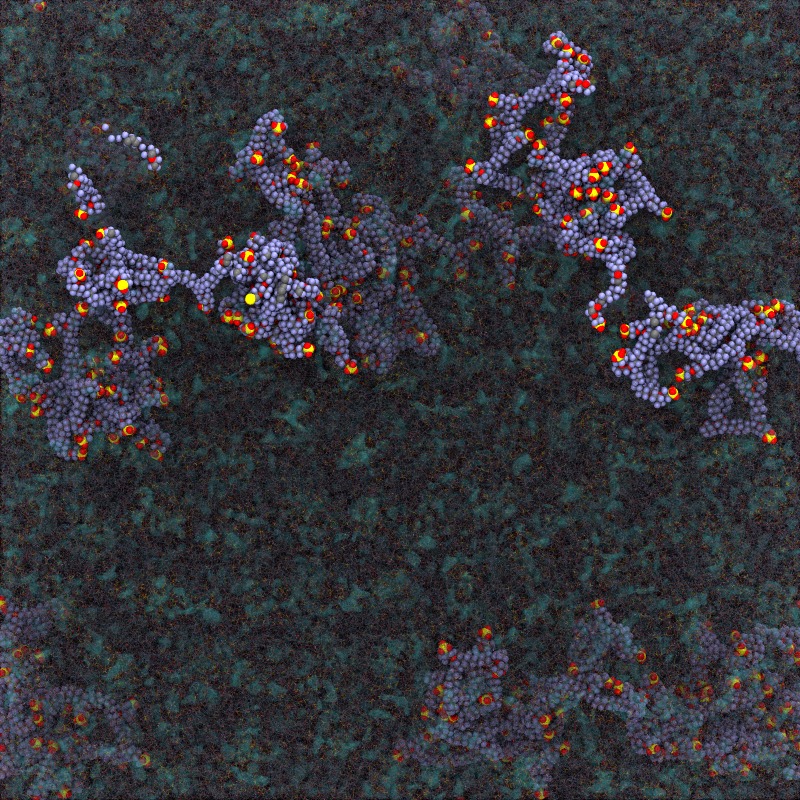 |
| Polysurfonic Acid (PSFA) polymer membrane has been simulated to understand the morphology change due to the water contents. Polymers with experimental lentghs were successfully simulated using multiscale modeling. Started with the chemically accurate coarse grain (CG) MD to obtain the relaxed membrane structure, and then all-atomic (AA) configuration was obtained by a reverse-mapping approach. Additional AA-MD under strain provides mechanical properties of PFSA membrane. The multiscale simulation technique is found to work pretty well for PSFA membranes. |
Electrolyte for Li-ion Battery
Molecular dynamics simulations have been used to investigate the structural and dynamical properties of ionic liquids. We are particuarlly interested in "solvate" ionic liquids, where a Lithium ion is strongly associated with a glyme molecule. The stable 1:1 Li-glyme complex weaken the interaction with counter ions (anions), and as a result, the salts solvated by glymes are in liquid state at ambient temperature. The stability of Li-glyme complex, however, is highly dependent on the choice of anion and the addition of the other organic solvents. In order to find a suitable combination of the component to make an ideal ionic liquids, many experimental and computational works have been done. A long-time MD simulation has also been used to characterize the ion dynamics. (e.g., see Shinoda et al. J. Chem. Phys. 2018)
 |
| A snapshot from MD simulation of 1:1 Li[TFSA]-Glyme4 system. Inset show a picture of typical Li(Glyme) complex found in the system. |
Further, sulfolane attracts attension as the other possible good solvent to make a useful elecrolyte for Li ion battery. In general, the physical properties including ion dynamics in electrolytes are altered by solvent concentration significantly, so that the molecular model (force field) should be useful over a wide range of solvent (or salt) concentration. A polarizable force field should be required to realize an accurate estimation of interaction among the molecules (ions and neutral solvents) over a range of solvent concentrations. We have developed a polarzable force field as an extension of OPLS-AA force field for the Li-anion salts as well as for sulfolane and its derivatives. MD simulations with the new force field produce molecular trajectories in which Li-ion dynamics is well reproduced, compared to the experimental diffusion coefficients and conductance. We applied this force field to a series of MD simulations of electrolytes including solvents like sulfolane. Analyses of dynamic correlation of Li-ions elucidate the feature or role of solvents in controlling the Li-ion dynamics.
